|
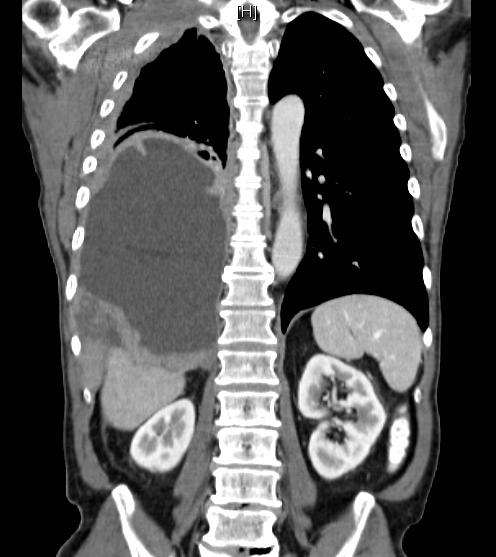
| Mésothéliome détecté dans la partie basse du poumon gauche. Source iconographique: https://en.wikipedia.org/wiki/Mesothelioma#/media/ |
Les options de traitement pour le mésothéliome pleural malin sont rares. Le tazemetostat, un activateur oral sélectif de l'inhibiteur de l'homologue 2 de Zeste 2 (EZH2), a montré une activité antitumorale dans plusieurs cancers hématologiques et tumeurs solides. Notre objectif était d'évaluer l'activité antitumorale et l'innocuité du tazemetostat chez les patients atteints d'un mésothéliome pleural malin récidivant ou réfractaire mesurable.
Nous avons mené une étude ouverte de phase 2 à un seul bras dans 16 hôpitaux en France, au Royaume-Uni et aux États-Unis. Les patients éligibles étaient âgés de 18 ans ou plus et avaient un mésothéliome pleural malin de toute histologie en rechute ou réfractaire après un traitement avec au moins un régime contenant du pemetrexed, un indice de performance Eastern Cooperative Oncology Group (ECOG) de 0 ou 1 et une espérance de vie supérieure à 3 mois. Dans la partie 1 de l'étude, les participants ont reçu 800 mg de tazemetostat par voie orale une fois le jour 1, puis deux fois par jour à partir du jour 2. Dans la partie 2, les participants ont reçu du tazemetostat oral 800 mg deux fois par jour à partir du jour 1 du cycle 1, en utilisant un plan de Green-Dahlberg en deux étapes. Le tazémétostat a été administré en cycles de 21 jours pendant environ 17 cycles. Le critère d'évaluation principal de la partie 1 était la pharmacocinétique du tazemetostat et de son métabolite au jour 15 après l'administration de 800 mg de tazemetostat, mesurée par la concentration sérique maximale (Cmax), le temps jusqu'à la Cmax (Tmax), l'aire sous la courbe concentration-temps (ASC) au jour 15 (ASC0–t), l'aire sous la courbe du temps 0 extrapolée à l'infini (ASC0–∞) et la demi-vie (t1/2) du tazemetostat, évaluées chez tous les patients inclus dans la partie 1. Le critère d'évaluation de la partie 2 était le taux de contrôle de la maladie (la proportion de patients avec une réponse complète, une réponse partielle ou une maladie stable) à la semaine 12 chez les patients atteints de mésothéliome pleural malin selon le protocole avec inactivation de BAP1 déterminée par immunohistochimie. La population de tolérance incluait tous les patients ayant eu au moins une évaluation de tolérance post-dose. Cet essai est maintenant terminé. (…).
Entre le 29 juillet 2016 et le 2 juin 2017, 74 patients ont été recrutés (13 dans la partie 1 et 61 dans la partie 2) et ont reçu du tazemetostat, dont 73 (99 %) avaient des tumeurs inactivées par BAP1. Dans la partie 1, après administration répétée de tazemetostat à l'état d'équilibre, au jour 15 du cycle 1, la Cmax moyenne était de 829 ng/mL (coefficient de variation de 56,3 %), la Tmax médiane était de 2 h (intervalle 1 à 4), l'ASC0–t moyenne était de 3310 h·ng/mL (coefficient de variation 50,4 %), l'ASC0–∞ moyenne était de 3180 h·ng/mL (46,6 %) et la moyenne géométrique t1/2 était de 3,1 h (13,9%). Après un suivi médian de 35,9 semaines (Intervalle InterquartiIe [IQR] 20,6–85,9), le taux de contrôle de la maladie dans la partie 2 chez les patients atteints de mésothéliome pleural malin inactivé par BAP1 était de 54 % (Intervalle de Confiance [IC] à 95 % 42–67 ; 33 de 61 patients) à la semaine 12. Aucun patient n'a eu de réponse complète confirmée. Deux patients ont eu une réponse partielle confirmée : l'un avait une réponse partielle en cours d'une durée de 18 semaines et l'autre avait une durée de 42 semaines. Les événements indésirables de grade 3-4 les plus courants liés au traitement étaient l'hyperglycémie (cinq [7 %] patients), l'hyponatrémie (cinq [7 %]) et l'anémie (quatre [5 %]) ; des événements indésirables graves ont été signalés chez 25 (34 %) des 74 patients. Cinq (7 %) des 74 patients sont décédés pendant l'étude ; aucun décès lié au traitement n'est survenu.
Un raffinement
supplémentaire des biomarqueurs de l'activité du tazemetostat dans le
mésothéliome pleural malin au-delà de l'inactivation de BAP1* pourrait aider à
identifier un sous-ensemble de tumeurs les plus susceptibles de tirer un
bénéfice prolongé ou une diminution de cette thérapie. Marjorie G Zauderer MD,
et al, dans The Lancet Oncology, publication en ligne en avant-première,
16 mai 2022
*La protéine BAP1 ou BRCA1 associated protein-1 (ubiquitin carboxy- terminal hydrolase) est une enzyme de dé-ubiquitination codée par le gène BAP1 situé sur le chromosome humain n°3 . BAP1 (BRCA1-associated protein 1) se lie au domaine N-terminal de BRCA1 et est considéré comme un gène suppresseur des tumeurs ( cf. www.diagomics.com )
Financement : Epizyme
Source : The Lancet Online / Préparation post :
NZ